





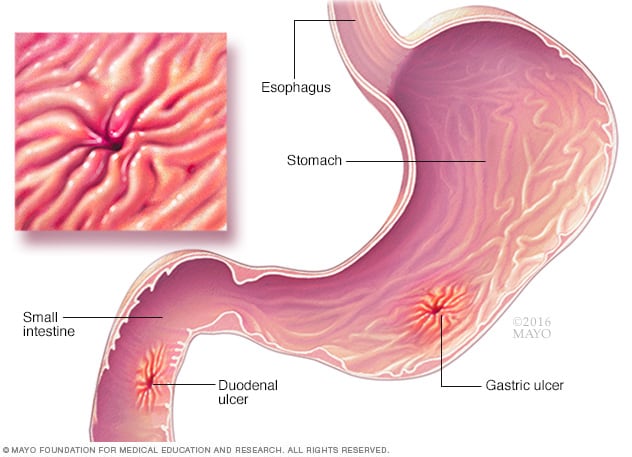
Cáncer colorrectal hereditario sin poliposis (HNPCC), también llamado Síndrome de Lynch, es una condición genética autosómica dominante que se asocia con un alto riesgo de cáncer de colon, así como con otros cánceres que incluyen cáncer de endometrio (el segundo más común), ovario, estómago, intestino delgado, tracto hepatobiliar, tracto urinario superior, cerebro y piel. El mayor riesgo de estos cánceres se debe a mutaciones heredadas que dificultan la reparación de los desajustes del ADN. Es un tipo de síndrome canceroso.
Síntomas del síndrome de Lynch
Riesgo de cáncer
Riesgo de por vida y edad media al momento del diagnóstico de los cánceres asociados al síndrome de Lynch
| Tipo de cancer | Riesgo de por vida (%) | Edad media al diagnóstico (años) |
| Colorrectal | 52-58 | 44-61 |
| Endometrial | 25-60 | 48-62 |
| Gástrico | 6-13 | 56 |
| Ovárico | 4-12 | 42,5 |
Además de los tipos de cáncer que se encuentran en el cuadro anterior, se entiende que el síndrome de Lynch también contribuye a un mayor riesgo de cáncer de intestino delgado, cáncer de páncreas, cáncer de uréter / pelvis renal, cáncer de vías biliares, cáncer de cerebro y neoplasias sebáceas. El aumento del riesgo de cáncer de próstata y cáncer de mama también se ha asociado con el síndrome de Lynch, aunque esta relación no se comprende del todo.
Dos tercios de los cánceres de colon ocurren en el colon proximal y los signos y síntomas comunes incluyen sangre en las heces, diarrea o estreñimiento y pérdida de peso involuntaria. La edad media de diagnóstico de cáncer colorrectal es de 44 años para los miembros de familias que cumplen con los criterios de Amsterdam. La edad promedio de diagnóstico del cáncer de endometrio es de aproximadamente 46 años. Entre las mujeres con HNPCC que tienen cáncer de colon y de endometrio, aproximadamente la mitad presenta primero cáncer de endometrio, lo que hace que el cáncer de endometrio sea el cáncer centinela más común en el síndrome de Lynch. El síntoma más común del cáncer de endometrio es el sangrado vaginal anormal. En el HNPCC, la edad media de diagnóstico del cáncer gástrico es de 56 años, siendo el adenocarcinoma de tipo intestinal la patología notificada con mayor frecuencia. Los cánceres de ovario asociados a HNPCC tienen una edad promedio de diagnóstico de 42,5 años; aproximadamente el 30% se diagnostica antes de los 40 años.
Se ha encontrado una variación significativa en la tasa de cáncer dependiendo de la mutación involucrada. Hasta la edad de 75 años, los riesgos de cáncer colorrectal, cáncer de endometrio, cáncer de ovario, cáncer gastrointestinal superior (gástrico, duodenal, de vías biliares o pancreático), del tracto urinario, cáncer de próstata y tumores cerebrales eran los siguientes: para las mutaciones MLH1, el riesgo fue del 46%, 43%, 10%, 21%, 8%, 17% y 1% respectivamente; para las mutaciones de MSH2, los riesgos fueron 57%, 17%, 10%, 25%, 32% y 5%, respectivamente; para las mutaciones de MSH6, los riesgos fueron del 15%, 46%, 13%, 7%, 11%, 18% y 1%, respectivamente.
| Gene | Riesgo de cáncer de ovario | Riesgo de cáncer de endometrio |
|---|---|---|
| MLH1 | 4-24% | 25-60% |
| MSH2 / EPCAM | 4-24% | 25-60% |
| MSH6 | 1-11% | 16-26% |
| PMS2 | 6% (riesgo combinado) | 15% |
Genética

El HNPCC se hereda de forma autosómica dominante. El sello distintivo de HNPCC es la reparación defectuosa del desajuste del ADN, que causa una tasa elevada de cambios de un solo nucleótido e inestabilidad de microsatélites, también conocida como MSI-H (la H es “alta”). La MSI es identificable en muestras de cáncer en el laboratorio de patología. La mayoría de los casos dan como resultado cambios en las longitudes de las repeticiones de dinucleótidos de las nucleobases citosina y adenina (secuencia: CACACACACA…).
Los 4 genes principales involucrados en HNPCC normalmente codifican proteínas que forman dímeros para funcionar:
- La proteína MLH1 se dimeriza con la proteína PMS2 para formar MutLα, que coordina la unión de otras proteínas involucradas en la reparación de errores de emparejamiento como la helicasa de ADN, la proteína de unión a ADN monocatenario (RPA) y las polimerasas de ADN.
- La proteína MSH2 se dimeriza con la proteína MSH6, que identifica los desajustes mediante un modelo de abrazadera deslizante, una proteína para buscar errores.
El deterioro de cualquiera de los genes para el dímero de la proteína afecta la función de la proteína. Estos 4 genes están involucrados en la corrección de errores (reparación de desajustes), por lo que la disfunción de los genes puede conducir a la incapacidad de corregir errores de replicación del ADN y causar HNPCC. Se sabe que el HNPCC está asociado con otras mutaciones en genes implicados en la vía de reparación de errores de apareamiento del ADN:
| Nombre OMIM | Genes implicados en HNPCC | Frecuencia de mutaciones en familias de HNPCC | Lugar | Primera publicacion |
|---|---|---|---|---|
| HNPCC1 (120435) | MSH2 / EPCAM | aproximadamente 60% | 2p22 | Fishel 1993 |
| HNPCC2 (609310) | MLH1 | aproximadamente 30% | 3p21 | Papadopoulos 1994 |
| HNPCC5 | MSH6 | 7-10% | 2p16 | Miyaki 1997 |
| HNPCC4 | PMS2 | relativamente poco frecuente | 7p22 | Nicolaides 1994 |
| HNPCC3 | PMS1 | Reporte de un caso | 2q31-q33 | Nicolaides 1994 |
| HNPCC6 | TGFBR2 | Reporte de un caso | 3p22 | |
| HNPCC7 | MLH3 | cuestionado | 14q24.3 |
La mayoría de las personas con HNPCC heredan la afección de uno de sus padres. Sin embargo, debido a la penetrancia incompleta, la edad variable del diagnóstico del cáncer, la reducción del riesgo de cáncer o la muerte temprana, no todas las personas con una mutación del gen HNPCC tienen un padre que haya tenido cáncer. Algunas personas desarrollan HNPCC de novo en una nueva generación, sin heredar el gen. A menudo, estas personas solo se identifican después de desarrollar un cáncer de colon en una etapa temprana. Los padres con HNPCC tienen un 50% de posibilidades de transmitir la mutación genética a cada hijo. También es importante señalar que la mutación deletérea en uno de los genes MMR por sí sola no es suficiente para causar cáncer, sino que es necesario que ocurran más mutaciones en otros genes supresores de tumores.
Diagnóstico del síndrome de Lynch
El diagnóstico de síndrome de Lynch se aplica a personas con una mutación del ADN de la línea germinal en uno de los genes MMR (MLH1, MSH2, MSH6 y PMS2) o el gen EPCAM, identificado mediante pruebas genéticas. Los candidatos para las pruebas genéticas de la línea germinal pueden identificarse mediante criterios clínicos como los Criterios Clínicos de Amsterdam y las Directrices Bethesda, o mediante el análisis de tumores mediante inmunohistoquímica (IHC) o pruebas de inestabilidad de microsatélites (MSI). En los EE. UU., Las sociedades profesionales recomiendan probar todos los cánceres de colon para detectar MSI o IHC como detección del síndrome de Lynch, pero esto no siempre se realiza debido a las limitaciones de costos y recursos. Las pruebas genéticas están disponibles comercialmente y consisten en un análisis de sangre.
Inmunohistoquímica
La inmunohistoquímica (IHC) es un método que se puede utilizar para detectar la expresión de la proteína de reparación anormal de errores de emparejamiento (MMR) en tumores asociados con el síndrome de Lynch. Si bien no es un diagnóstico de síndrome de Lynch, puede desempeñar un papel en la identificación de las personas que deberían someterse a una prueba de línea germinal. Dos métodos de implementación de las pruebas IHC incluyen pruebas basadas en la edad y pruebas universales para todas las personas. Actualmente, no existe un acuerdo generalizado sobre qué método de detección se debe utilizar. Las pruebas de IHC basadas en la edad se han sugerido en parte debido a los análisis de costo-beneficio, mientras que las pruebas universales para todas las personas con cáncer colorrectal aseguran que las personas con síndrome de Lynch no se pierdan. Para abordar los costos, los investigadores están tratando de predecir la MSI o IHC directamente a partir de la apariencia del tumor bajo el microscopio, sin realizar ninguna prueba molecular.
Inestabilidad de microsatélites
Las mutaciones en los sistemas de reparación de errores de emparejamiento del ADN pueden provocar dificultades para transmitir regiones dentro del ADN que contienen patrones repetidos de dos o tres nucleótidos (microsatélites), también conocido como inestabilidad de microsatélites (MSI). La MSI se identifica mediante la extracción de ADN tanto de una muestra de tejido tumoral como de una muestra de tejido normal, seguida de un análisis por PCR de las regiones de microsatélites. El análisis de MSI se puede utilizar para identificar a las personas que pueden tener síndrome de Lynch y dirigirlas para que se realicen más pruebas.
Clasificación
Tres grupos principales de cánceres MSI-H (inestabilidad de microsatélites – MSI) pueden reconocerse mediante criterios histopatológicos:
- cánceres del lado derecho poco diferenciados
- cánceres mucinosos del lado derecho
- adenocarcinomas en cualquier ubicación que muestren cualquier nivel medible de linfocitos intraepiteliales (TIL)
Los criterios histopatológicos no son lo suficientemente sensibles para detectar la MSI a partir de la histología, pero los investigadores están tratando de utilizar la inteligencia artificial para predecir la MSI a partir de la histología.
Además, el HNPCC se puede dividir en síndrome de Lynch I (cáncer de colon familiar) y síndrome de Lynch II (HNPCC asociado con otros cánceres del tracto gastrointestinal o del sistema reproductivo).
Prevención
Prueba de pantalla
Se recomiendan el asesoramiento genético y las pruebas genéticas para las familias que cumplen con los criterios de Amsterdam, preferiblemente antes de la aparición del cáncer de colon.
Cáncer de colon
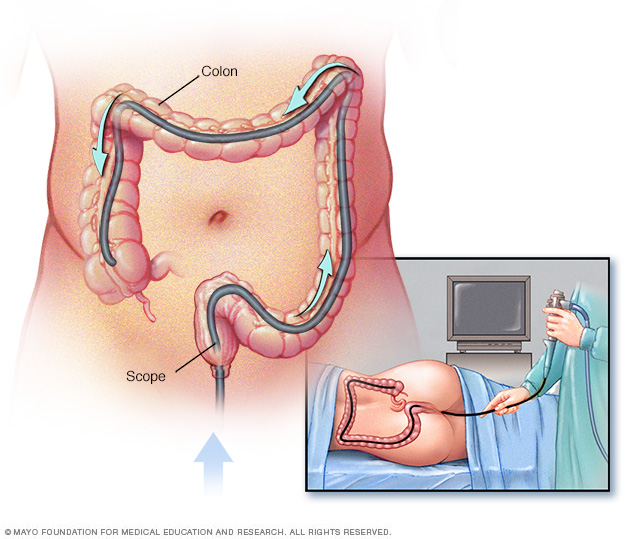
Las colonoscopias se recomiendan como método preventivo de vigilancia para las personas que tienen el síndrome de Lynch o genes asociados con el LS. Específicamente, se recomienda que las colonoscopias comiencen a los 20-25 años para los portadores de mutaciones MLH1 y MSH2 y a los 35 años para los portadores de mutaciones MSH6 y PMS2. Luego, se debe realizar una vigilancia colonoscópica en un intervalo de 1 a 2 años para los pacientes con síndrome de Lynch.
Cáncer de endometrio / ovario
Se recomienda una ecografía transvaginal con o sin biopsia de endometrio anualmente para la detección del cáncer de ovario y endometrio. Para las mujeres con síndrome de Lynch, se puede utilizar un análisis de sangre CA-125 anual para detectar el cáncer de ovario; sin embargo, existen datos limitados sobre la eficacia de esta prueba para reducir la mortalidad.
Otros cánceres
También existen estrategias para detectar otros cánceres en forma temprana o reducir las posibilidades de desarrollarlos que las personas con síndrome de Lynch pueden discutir con su médico, sin embargo, su efectividad no está clara. Estas opciones incluyen:
- Endoscopias superiores para detectar cáncer de estómago e intestino delgado cada 3 a 5 años, comenzando a los 30 años como mínimo (preferiblemente en un entorno de investigación)
- Análisis de orina anual para detectar cáncer de vejiga, a partir de los 30 años como mínimo (preferiblemente en un entorno de investigación)
- Exámenes físicos y neurológicos anuales para detectar cáncer en el sistema nervioso central (cerebro o médula espinal), a partir de los 25 años como mínimo
Criterios de Amsterdam
Los siguientes son los criterios de Amsterdam para identificar candidatos de alto riesgo para las pruebas genéticas moleculares:
Criterios Amsterdam I (deben cumplirse todos los puntos):
- Tres o más miembros de la familia con un diagnóstico confirmado de cáncer colorrectal, uno de los cuales es pariente de primer grado (padre, hijo, hermano) de los otros dos
- Dos generaciones sucesivas afectadas
- Uno o más cánceres de colon diagnosticados antes de los 50 años
- Se ha excluido la poliposis adenomatosa familiar (PAF).
Los criterios de Amsterdam II se desarrollaron en 1999 y mejoraron la sensibilidad diagnóstica del síndrome de Lynch al incluir cánceres de endometrio, intestino delgado, uréter y pelvis renal.
Amsterdam Criteria II (se deben cumplir todas las viñetas):
- Tres o más familiares con cánceres relacionados con HNPCC, uno de los cuales es pariente de primer grado de los otros dos
- Dos generaciones sucesivas afectadas
- Uno o más de los cánceres relacionados con HNPCC diagnosticados antes de los 50 años
- Se ha excluido la poliposis adenomatosa familiar (PAF).
Cirugía
La histerectomía profiláctica y la salpingooforectomía (extirpación del útero, las trompas de Falopio y los ovarios para prevenir el desarrollo del cáncer) se pueden realizar antes de que se desarrolle el cáncer de ovario o endometrio.
Tratamiento del síndrome de Lynch
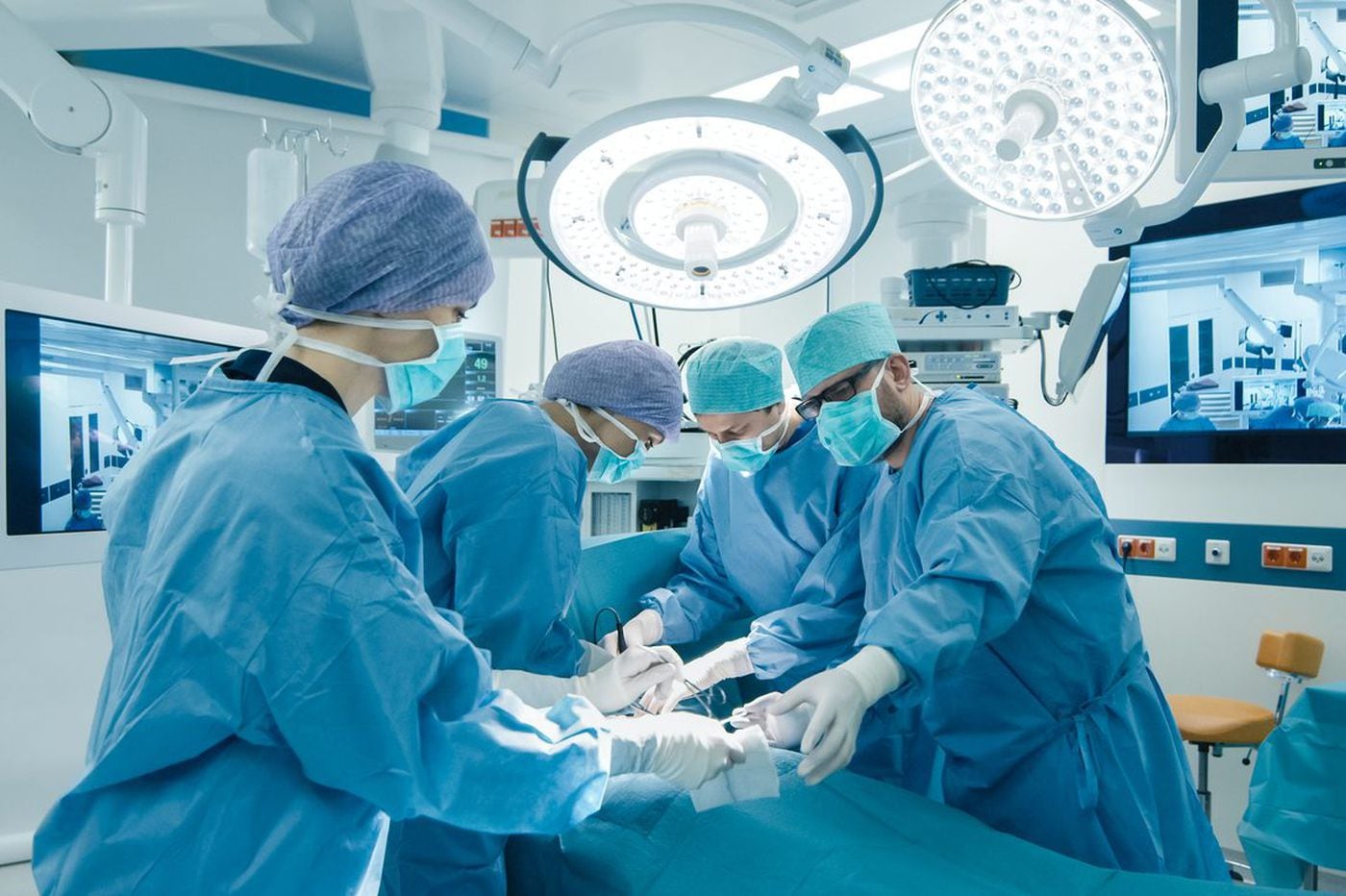
La cirugía sigue siendo la terapia de primera línea para HNPCC. Los pacientes con síndrome de Lynch que desarrollan cáncer colorrectal pueden ser tratados con colectomía parcial o colectomía total con anastomosis ileorrectal. Debido al mayor riesgo de cáncer colorrectal después de una colectomía parcial y una calidad de vida similar después de ambas cirugías, una colectomía total puede ser un tratamiento preferido para el HNPCC, especialmente en pacientes más jóvenes.
Existe una controversia en curso sobre el beneficio de las terapias adyuvantes basadas en 5-fluorouracilo para los tumores colorrectales relacionados con el HNPCC, en particular los que se encuentran en los estadios I y II.
- La terapia con anticuerpos anti-PD-1 puede ser eficaz.
El bloqueo de puntos de control con terapia anti-PD-1 es ahora la terapia de primera línea preferida para el cáncer colorrectal avanzado con Inestabilidad de microsatélites avanzada.
.